A Nextflow pipeline for processing RNASeq sequencing data.
The pipeline was written by The Bioinformatics & Biostatistics Group at The Francis Crick Institute, London.
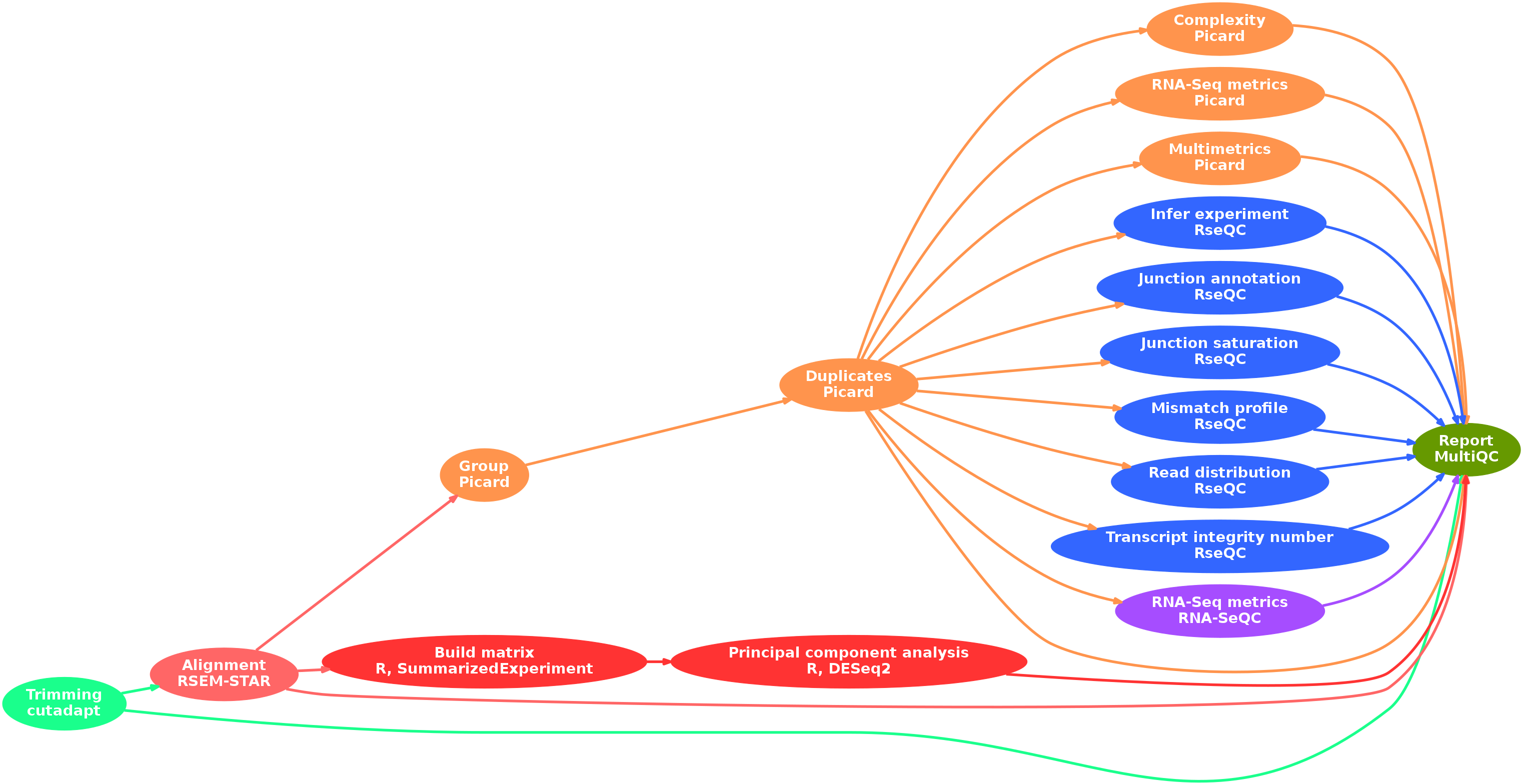
- Raw read QC (
FastQC) - Adapter trimming (
cutadapt) - Alignment and quantification (
RSEM,STAR) - Sorting and indexing (
SAMtools) - Quality control metrics:
picard:- Groups (
AddOrReplaceReadGroups) - Duplicates (
MarkDuplicates) - Library complexity (
EstimateLibraryComplexity) - Various metrics (
CollectRnaSeqMetrics,CollectMultipleMetrics)
- Groups (
RSeQC:- Samples quality (
infer_experiment.py,read_distribution.py,tin.py) - Alternative splicing (
junction_annotation.py,junction_saturation.py) - Mismatch (
mismatch_profile.py)
- Samples quality (
RNA-SeQC
- Preparation for statistical analysis:
- Create a count matrix (
R,SummarizedExperiment) - Perform a principal component analysis (
R,DESeq2)
- Create a count matrix (
- Collect and present a report (
MultiQC)
The documentation for the pipeline can be found in the docs/ directory:
- Installation
- Design file
- Pipeline configuration
- Running the pipeline
- Output and interpretation of results
- Troubleshooting
The pipeline was written by the The Bioinformatics & Biostatistics Group at The Francis Crick Institute, London.
The pipeline was developed by Gavin Kelly, Harshil Patel, Nourdine Bah and Philip East.
This project is licensed under the MIT License - see the LICENSE.md file for details.